
�������: 1-15 ���鵽���ṹ��ѧ ̼����ؼ�¼40�� . ��ѯʱ��(0.919 ��)

��������ѧ������ѧѧ�����桿����Ԫ���ϵĽṹɸѡ����-̼��ϵ�IJ���Ԥ��

���Ϲ�LED��һ����Ҫ�Ĺ���������ڹ��������������ݴ��桢��ʾ����ͶӰ��Ӧ��������о��Ӧ��DZ������������ɫLED��Ҫ��III-V�������뵼��Ϊ����㣬��ʹ�õĽ���-�л���ѧ������������ɱ��ߺ��ո��ӣ�̽�������������۵����Ϲ�LED����������������Ҫ�о�����
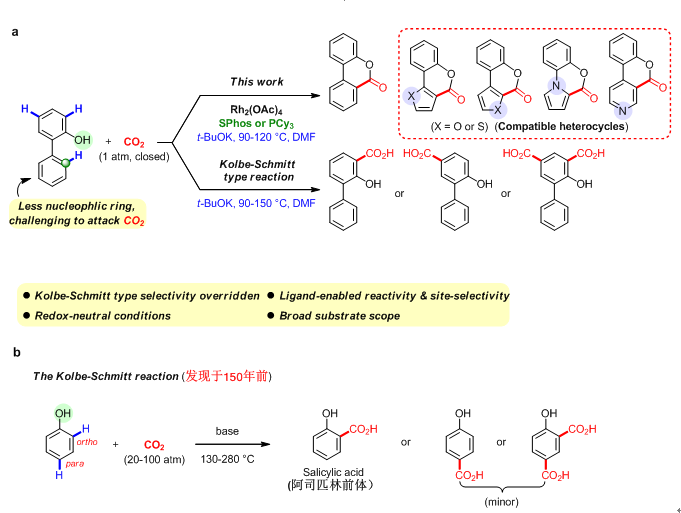
�й���ѧԺ�������ʽṹ�о���������̼�����ѡ����C�CH���Ȼ�����Ӧ�о����չ��ͼ��
�й���ѧԺ�������ʽṹ�о��� ������̼ ѡ���� C�CH���Ȼ�����Ӧ
2018/6/8
�й���ѧԺ�������ʽṹ�о����ṹ��ѧ�����ص�ʵ���Һ��п�Ժú���Ҷ�������ؼ����ص�ʵ�����о�Ա��ٿ����飬�ڹ��ҡ�����ǧ�˼ƻ������п�Ժս�����ȵ��Ƽ�ר�B�ࣩ��������Ȼ��ѧ���𡢸���ʡ���˼ƻ��Լ�����ʡ��Ȼ��ѧ����������£�ʹ�ù��ɽ���Rh(II)��Ϊ������������ĵ��������£��ɹ�ʵ����������ԭ���������µ�CO2�����2-�������������Ķ��Է���C�CH����λ��ѡ�����Ȼ�����Ӧ��ͼ1a������...

�й���ѧԺ�������ʽṹ�о����ڽ����ӻ�߲�������ƺϳ�������ԭ������̼�о��л��չ��ͼ��
�й���ѧԺ�������ʽṹ�о��� �����ӻ�߲�� ������ �����ԭ ������̼
2017/12/26
�й���ѧԺ�������ʽṹ�о����ṹ��ѧ�����ص�ʵ���������տ��������Ž���������������÷ӻ�߲����Ϊ���壬��ƺϳ���һϵ�о���������(nbo)��������ṹ�ķӻ�߲��MOFs��������ϵ��ȶ��Էdz��ã���������0.1M������ˮ��Һ�����ٿ����ȶ�7�죬�����ڱ��͵���������ˮ��Һ��~20M�������ٿ����ȶ�7�졣������߲�����п������벻ͬ�Ľ���λ����Ϊ���������ģ����ǶԽ������ķӻ�߲��MOFs�����˹��CO...
����̼�����������ܼ�������̽��Ļ������-���䶯��ѧģ��
����̽�� �������-���䶯��ѧ ����̼���� ��ԥʱ�� ��Ƶ��λ��
2010/3/9
���û������-���䶯��ѧģ�ⷽ��, �����˲�ͬ�ܾ��ĵ���̼����(SWCT)�������ܼ�벵ľ���ֲ��Լ�����I2���ӵ���ԥ����ѧ, ������I2���ӵ���Ƶ��λ�ơ���ԥʱ��������̼���ܹܾ��ߴ�仯�Ĺ�ϵ. ��I2���ӵ���Ƶ��λ��Ϊ̽��, ����I2��������Χ�������õ�ʵʱ��Ϣ, �����˹ܱڡ������ܼ��Թ���̽��Ĺ���, ��ԭ�ӡ����Ӳ���Ͻ�ʾ���յ�Ƶ��λ�Ƶ��ۻ���; ����, ͨ������̽�����...
������ϵ����ӻ�ѧONIOM(B3LYP/6-311++G**:UFF)����, �о��˲�ֱͬ���ķ�������(CNT(5,5)��CNT(6,6)��CNT(8,8))�;����(CNT(9,0)��CNT(10,0)��CNT(11,0))����̼����(CNTs)���������ö�����������ӽṹ���ȽⷴӦ��Ӱ��. ���ӽṹ��������, �뵥�������������, ������ֱ����С��CNT(5,5)��CNT(9,0)̼����...
̼���������ߵ��Ʊ��ͽṹ
̼����(CNTs) ̼���������� ����
2009/12/28
��̼����Ϊģ�壬ͨ������̼��������۵Ļ�������˱�ֱ����̼������.�������ߵĽṹ�ͳɷֽ����о������������������ҪΪB4C������.�ڲ���B4C�����ߵĶ˲�����Ni��������Щ�˲�����Ni�����������߹��������״���.������B4C�����ߵ��������ƣ�B4C�����ߵ�������ҪΪ��ԭ����̼��������ɢ��������ѧ��Ӧ��ʹ��̼���ܾ���ṹ�������飬�γ�B4C������.��Ӧ����ԭ�Ӳ���ȡ����̼������̼ԭ��...
�ɹ������ϳɵ���ɢ����������̼�Ļ�ѧ��̬��
����̼Ԫ�شغ���ijɼ�����
̼Ԫ�ش� ������ Boys���� ��ѧ��
2009/12/11
��ab initio 3-21Gˮƽ��, �������ݶȷ��Ż�������̼Ԫ�شغ���C_n~e(nΪ�ɴ�ԭ�Ӹ���, eΪ���)��ƽ�⼸�νṹ. ���õĵ�������ɴ�ԭ�Ӹ����ĸı�, ���ֳ���ͬ�̶ȵ���ż����仯����. ��ab initio���������, ��Boys����, ����ռ��������ӹ�����ж��任, �õ������ǵĶ�����ӹ��. �Զ�����ӹ�����ʵķ�������, ����̼Ԫ�شغ�����, ��Ҫ������˫���ĦҺ�...
���ھ����ܶȷ�������, ���õ�һԭ������, ������(5, 5)�ͺ�(8, 0)������̼���ܵ�ԭ��ģ��, ���������˿ڽ�֦1��8���ǻ�������, ����DMol3��BLYP��������ṹ�����Ż�, ������CASTEP������������ӷֲ���̬�ܶȵı仯, �Ӷ������ǻ������Ŷ�̼���ܵ��ӽṹ�͵����������Ե�Ӱ��. �������, ��֦�ǻ���̼���ܵĵ��ӽṹ���Ըı�, �����ܼ��ϵĵ���̬�ܶ��½�, ���ռ�ݹ�...
����ONIOM(B3LYP/6-31+G*: UFF)������ż������������ʽ����̼����CNT(8,8)�ڵĽṹ�����ӹ�������˳���칹������������˼���. �������, ��ʽż��������CNT(8,8)̼��������һ�����ȹ���. ����̼���ܵ���������, ż�������ӵı�����CN����һ������ת, ��ʽż������ƽ��ṹ������Ť��, �������ṹ�����仯������. ������̼�����ڵ�ż������˳���칹���������...
�����ܶȷ�������(DFT), ��B3LYP/6-311+G**ˮƽ��, �о����������ƽ������λ̼ԭ��(ptC)��ƽ������λ̼ԭ��(ppC)����̼������. ���������ͻ���������C3B2H4(����ptC)��CB4H2(����ptC)��CB5H2(����ppC)�����ȶ��ṹ�͡�CHCH����Ԫ�����������õ���. ��������̽������Щ���͵���̼������ijɼ�����, ���������Լ�������. �о��������: ����ptC...
�������þ��䶯��ѧģ��, �����׳߶��϶Ե;���ϩ��(PE)�ں������ʽ��CNTs�ϵ������������о�. ģ�����������, PE����CNTs����������γɵĹ���������CNTs�ijߴ�ƥ����кܴ��ϵ, ����PE����CNTs�ijߴ���ƥ��ʱ, �����γɹ�����ͼ��������, ��Ϊ���õ�����װ�ṹ. ������׳߶ȵ�ͼ�������ƺ�������.
ԭ�ӵ�������ʸ��������Ǻ˴Ź���̼��ѧλ�Ƽ���
13C�˴Ź���ѧλ�ƣ���ǣ�ԭ�ӵ�������ʸ������Ԫ���Իع飻��������ϵ
2009/11/18
�ӷ��Ӷ�ά���˽ṹ������ ������������ԭ�����������ۻ�ѧ������ԭ�ӵ�������ʸ��(Atomic Elementary Electronegativity Interaction Vector, AEIV)��25������й���148���ȼ۹���̼ԭ�ӽ����˱��������Դ˽���������ģ�ⵥ�Ƿ���13C NMR��ѧλ�Ƶ���������Ԫ���Իع鷽�̣� ����ģ�͵ĸ����ϵ��Rcum���������QLOO����������R...
����ϩ����̼���ܲ�������Ķ���ѧģ���о�
̼���� ����ϩ ���� ����ѧģ��
2009/11/16
���þ���ķ��Ӷ���ѧģ�ⷽ���Ծ���ϩ(PE)���������ֲ�ͬ���͵�̼����(CNT)�е������������о�. ����������֮�����ɢϵ�����������; ����PE��������Ťת�Ƿֲ���ȡ�������PE����������˷���. �������, PE��������CNT�Ϻܺõ�����, ��PE�Ĺ��������λ����Ҫ���¶Ⱥ�CNT�İ뾶�й�, ��ܵ�����ϵ����.
�й��о����������а�-��
- ���ڼ���...
�й�ѧ���ڿ����а�-��
- ���ڼ���...
�����ѧ���л������а�-��
- ���ڼ���...
�й���ѧ���а�-��
- ���ڼ���...
�ˡ���-ƪ
- ���ڼ���...
�Ρ���-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
�������� -ƪ
- ���ڼ���...
֪ʶҪ��-ƪ
- ���ڼ���...
���ʶ�̬-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
ѧ��ָ��-ƪ
- ���ڼ���...
ѧ��վ��-ƪ
- ���ڼ���...