
�������: 1-15 ���鵽��������ѧ ��������ؼ�¼42�� . ��ѯʱ��(0.142 ��)
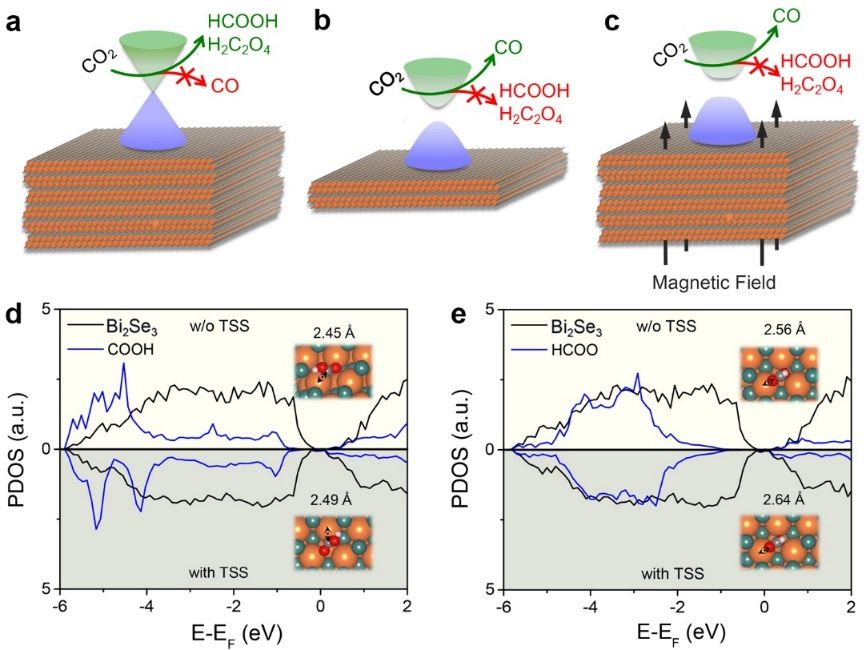
���գ��й���ѧ������ѧ���ܽ����ŶӺ������ɽ����ŶӺ���������������Ӵ��¸���о���Աͨ�������ʵ����ƣ�������������ʱ�䷴�ݶԳ��ԣ�Ϊ��ʾ����Ӧ�е���������̬�����ء�ЧӦ�ṩ��ȷ���ʵ��֤�ݡ���سɹ��ԡ�Experimental Demonstration of Topological Catalysis for CO2Electroreduction��Ϊ�ⷢ���ڡ�������ѧ�ᡷ�ڿ���J. A...

�й���ѧԺ�������ϼ����빤���о������������������ӻ�ѧ���۽��ʹ�������Դ������չ��ͼ��
�������ӻ�ѧ ������ ��Դ
2022/9/16
�Դ�1925��H.S Taylor������������ĵĸ���������������ͼͨ�����ַ�����������������Ե���Դ�������ܹ����ٶ�ȷ��Ԥ��������ĵ�λ�ã����ﵽ��Ƹ��Դ����ϵ�Ŀ�ġ������У������м�������/�Ѹ��Լ�d�����ĵ����۵��ܶȷ������ۼ���ȡ���˾�ijɹ����������ַ�������������������������˼��ߵ�Ҫ����Ҳ���ѶԲ��ϵ����о�������ȫ��ķ�������������������²��ϵ�Ԥ��ܡ���ô���Ƿ�...

��������ѧԺԺ��������������ѧ����ѧԺ���Ӳ��Ͽ�ѧ���ļ�ˬ�������߷��ֵ�һ���ɻ�ѧ�����������������䣨ͼ��
������ѧ����ѧԺ���Ӳ��Ͽ�ѧ���� ��ˬ ��ѧ������ �������
2020/7/10
���գ�������ѧ����ѧԺ���Ӳ��Ͽ�ѧ���IJ�ʿ�о�������Է����ˬ������������߷��ֵ����˰����VAl3�е��������ӿ��Բ��ܽϴ�Ļ�ѧ�ɷֱ仯��Ӱ�졣�о����֣���VAl3�д�����Vԭ�ӱ�Tiȡ��ʱ���侧���������˺ܴ�仯��Ȼ����ѧ���ʱ��ֲ��䣬˵�����еĵ����˵��Ӿ��и߶ȵ�³���ԡ�ֱ������35%��Vԭ�ӱ�ȡ�������ֵ����˰������ת��Ϊ����ƽӹ�Ľ�������ϵ�ѧ����������ṹ�������ܴ��ṹ����ͷ��ӹ��...

�й���ѧԺ�����о���ʵ�ֻ�������ת�����ϵĵ����ͻ������ܣ�ͼ��
�й���ѧԺ�����о��� ����ת������ ����� ͻ�������
2019/4/29
�й���ѧԺ�����о���/��������̬���������о����Ĺ������ص�ʵ�����о�Ա�������п�ԺԺʿ������쵼��L03��һֱ�����ڼ�����������ӷ����Ʊ��������ĸ��ѿ����ʽἰ�����Ե��ص��о����ÿ����鸱�о�Ա��衵�ͨ������Һ�����LSMO��Ĥʵ�����ĸ��������ϵĵ���仯�����������Һ���е�ˮ�����������绯ѧ���ӽ�����������Ҫ���ã�Adv. Mater. Interfaces 2, 1500407 (2015...
OCS ��ϩ��Ȳ��֮��S�������õĵ����ܶ���������
���Ӽ������ ���Ӿ����� �е����ܶȷֲ����� �����ܶ����˷���
2013/11/4
����B3LYP/6-311++G(d,p), B3LYP/aug-cc-pVDZ, MP2/6-311++G(d,p), MP2/aug-cc- pVDZ, MP2/aug-cc-pVTZ 5�ַ���, ������̼(OCS)�����е���ԭ����ϵ��ϩ��Ȳ��֮���γɵ�T ����������������о�, ���شӵ����ܶ����������ĽǶ�, ��OCS ��ϵ��ϩ��Ȳ��֮���γ�S�������ý����о�. ͨ����-�з���, �״λ�...
�л���Ⱦ������︻������������ָ������ѧģ��
�л���Ⱦ�� ���︻������ ��������ָ�� ��������ָ̬�� ���Ծ���ʸ�� �յ����ϻ��� ������Ч��ϵ(QSAR)
2009/12/28
���ݼ�������ָ��(xi)����������ָ̬��(ej)�����Ծ���ʸ��(mk)����239���л���Ⱦ�����︻������(FBC)��6����QSFR(�����ṹ-���︻�����������)ģ��, ������س̶ȸ�, ���������Ա�������. ��ģ�͵Ĵ�ͳ���ϵ��(R2)Ϊ0.821, LOO(levae-one-out)������֤ϵ��(Q2)Ϊ0.809, ֤���������õ��Ƚ��Լ�Ԥ������. ���ݽ����ģ�͵�6��������֪, Ӱ���л�...
ԭ�����͵�����״ָ̬��Ԥ�����������������Ŀ�����
ԭ�����͵�����״ָ̬�� ��������������� ���� ������Ч��ϵ ƫ��С����ģ��
2009/12/28
����ԭ�����͵�����״ָ̬��(ETSIAT)��17�����������������Ŀ������Խ��ж�����Ч��ϵ(QSAR)���о�. ���ع����ɸѡ, �õ�����4��ETSIAT����������ƫ��С����ģ��, �临����ϵ��R2����һ��������֤������ϵ��Q2�;��������RMSEE�ֱ�Ϊ0.806��0.736 ��0.248. �����������Ϊѵ������Ԥ�⼯��, ������ͬ������϶�ģ�ͽ����ⲿ��֤, �����ʾģ�;��нϸߵ��ⲿ...
��EHCO-ASED����, �Թ������Ȳ�ȾۺϷ�ӦMDA��PBT��PDA������(R=Li��CH_3��F)Ч��������о����������, ��ͬ����Ķ���Ȳ��ϵ��MDA��PBT��PDA�仯��,�ڷ�Ӧ����0.86����, ��ϵ����϶���ﵽ������ļ�Сֵ, ͬʱǰ�߹���Գ��Է���ͻ�䡣�����仯ʹ�ô˾ۺϷ�Ӧ��Ϊ�������ġ�����ı仯, �Ը��෴Ӧ�ķ�Ӧ���������Ӱ���С, ���Բ���PDA������ѧ�ȶ��Ե�Ӱ����...
�����л��ᱣ��ָ��������ָ����ԭ�����͵�����ָ��ģ��
�ǻ��� ������ ����ָ�� ����ָ�� ԭ�����͵�����ָ�� ��Ԫ�ع����
2009/12/3
��������̼ԭ�ӵ�ȡ�����������, �����������ָ��(wj), ���ԭ�����͵�����ָ��(En)�о�18�������ǻ���Ͱ�����ı���ɫ�ױ���ָ��(RM)�Ķ�����Ч��ϵ(QSRR). ����ѱ����Ӽ��ع齨�������Ԫ��ѧģ��, ��ͳ�����ϵ��(R2)Ϊ0.969����һ����(LOO)�Ľ�����֤ϵ��(Q2)Ϊ0.943, ���֤���������õ��Ƚ��Լ�Ԥ������. ���ݽ����ģ�͵�4���ṹ����(wj, E13, E...
���ڽṹ��Ϣ��ֵ���ĵ縺������ָ��������
��QCISD(T)/6-311++G(d,p)��B3LYP/6-311++G(d,p)�������о���HNCS��Clԭ�ӵķ�Ӧ����. ��Ӧ�þ������̬���ۺ������ֹ���̬���۽��С��������ЧӦ, ������200-2500 K�¶ȷ�Χ�ڸ���Ӧͨ�������ʳ���. �������, HNCS��Clԭ�ӷ�Ӧ����3����Ӧͨ��. ���¶ȵ���294 Kʱ, ����HCl+NCS�Ķ��ⷴӦ(a)������ͨ��, �¶ȸ���294 Kʱ...
5,7��-(�Ǽװ���)-��-8-�ǻ������������л��������ܶȷ�������Ȼ������������ܶ����������о�
8-�ǻ���������� ��� ��Ȼ��������� �����ܶ����˷��� ��ʱ�ܶȷ��� ����
2009/11/25
�����ܶȷ�������, ��B3LYP/6-31Gˮƽ�϶�5,7��-(�Ǽװ���)-��-�ǻ��������3�ֽ���M(M=Zn, Mg, Be)�л������M(5,7��-Iminomethylq2)2�Ľṹ������ȫ�Ż�, ����TDDFT�������������չ���. ͬʱ, ������Ȼ���������(NBO)�͵����ܶ���������(AIM)�����Է�������������˷���. �������, ������ֵ��ʵ��ֵ��������, �����������нϴ��...
CH3SH��HOO���������������Ľṹ��������ܶ���������
���� ��������ɻ� ��������� ��Ȼ��������� �����ܶ���������
2009/11/25
��DFT-B3LYP/6-311++G**ˮƽ�����CH3SH��HOO�������������ϵ��ȶ�����. ����������, ��HOO����O8��H7��Ϊ���ӹ�����CH3SH�����е�S5ԭ��Ϊ���������γɵ����������1��2��, O8��H7���Ա���������, ����������Ƶ�ʷ��������ĺ���, ����ֵ�ֱ�Ϊ330.1��320.4 cm-1; ��CH3SH��������S5��H6��Ϊ���ӹ�����HOO�Ķ˻�O9ԭ��Ϊ����������...
���Ȼ�п������Ϊԭ��, �����һ�����(DETA)Ϊģ���, ͨ��ˮ�ȷ��ϳ���һ�־��пտ��Ǽܽṹ���л�-���ӻ�����п������. ����ṹ�����������, �û����������ķ���ϵ, P42bc�ռ�Ⱥ, ��������a=b=1.46850(2) nm, c=0.89274(2) nm, ��=��=��=90��, V=1.92519(6) nm3, Z=8, Dc=2.641 g/cm3. �����������IR����...
��ԭ�����͵�������ָ̬����Ek�����������������ķ��ӽṹ.ͨ����Ԫ���Իع鼰��ѱ����Ӽ�������������������ײ���ж�(lg H50)���������ָ̬������Ԫ�������ģ��(QSPR)�������ϵ��RΪ0.930�����Ʊ����sΪ0.356.��Jacknnife���飬��ģ�;������õ��ȶ�����Ԥ������.Ԥ������ʾӰ�����������ײ���жȵ���Ҫ�ṹ����Ϊ�����ӻ��š�CH2������O������NH2�Լ������ӻ���=O��...
�й��о����������а�-��
- ���ڼ���...
�й�ѧ���ڿ����а�-��
- ���ڼ���...
�����ѧ���л������а�-��
- ���ڼ���...
�й���ѧ���а�-��
- ���ڼ���...
�ˡ���-ƪ
- ���ڼ���...
�Ρ���-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
�������� -ƪ
- ���ڼ���...
֪ʶҪ��-ƪ
- ���ڼ���...
���ʶ�̬-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
ѧ��ָ��-ƪ
- ���ڼ���...
ѧ��վ��-ƪ
- ���ڼ���...